

global and local Sequence Alignment
Oftentimes biologists will compare sequences of DNA or RNA to determine how similar they are. This seemingly simple task can unlock many of the secrets of life. For example, all species have common ancestors that can be identified through the comparison of DNA sequences. This can help biologists create better evolutionary trees. In addition, it is possible to identify regions of DNA that have been conserved (not changed) from species to species. These regions are likely essential for the function of the organism. Biologists use this data to ask questions like what is the purpose of this section of DNA? is it an exon (protein-coding region) or is it a regulatory region? what protein does it code? The parts of the DNA that change between species are also important to identify. They can help biologists figure out which mutations happened to get from one strand to another.
The two main types of sequence alignment are Global Alignment and Local Alignment. Global Alignment involves aligning sequences end to end. Which is what sounds like, the output is two sequences that are the same length. On the other hand, Local Alignment is aligning a portion of one sequence to a portion of another sequence. The point of local alignment is to find two subsequences (portions of the main sequence) that are very similar. This is useful in cases like finding a specific gene in a chromosome or a sequence motif in two very different sequences. Although there are specific algorithms for finding sequence motifs (parts of the DNA sequence that follow a specific structure and are likely functional). Global alignment can be useful in phylogenetics for determining mutations between closely related DNA strands. It is also desired for algorithms to be able to deal with gaps which are places in the alignment where one strand is missing a base. These are indicative an insertion mutation which when a base is inserted accidently. In the sample alignment below the gaps are denoted by a "-" character.
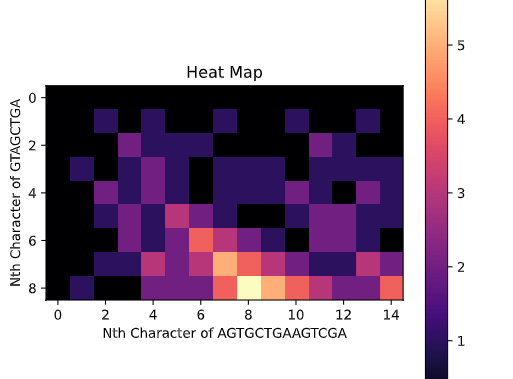
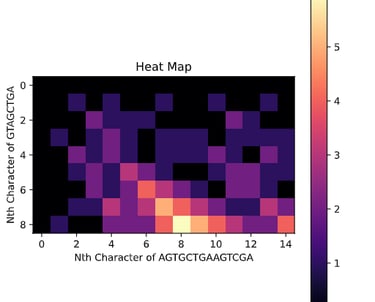
This task has been optimized for computers through several algorithms devised to solve the sequence alignment problem. I implemented two of these algorithms, the Needleman-Wunsch algorithm for Global Alignment and the Smith-Waterman algorithm for Local Alignment with Python using NumPy. I also created a GUI so that users can easily enter in two sequences and get an alignment as well as view a heat map showing how the algorithm found the alignment. I also included the ability to edit the scoring matrix which changes how the algorithm decides upon the best alignment.
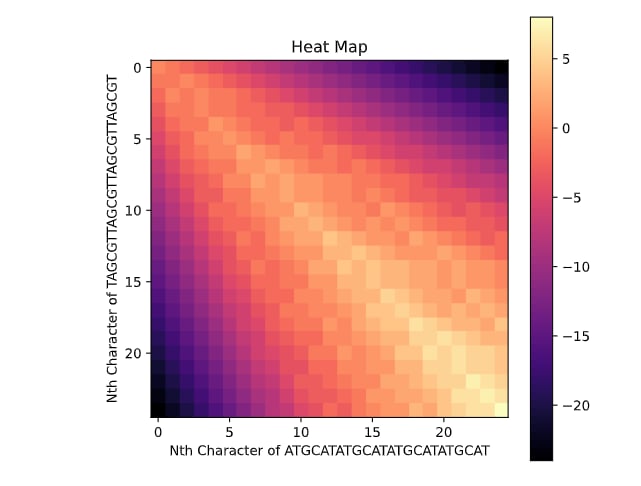
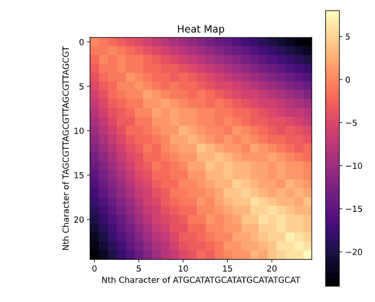

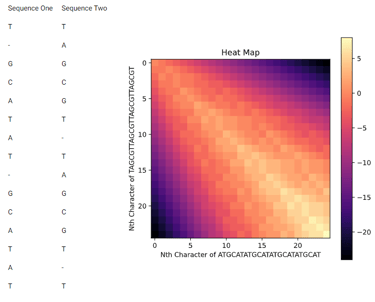
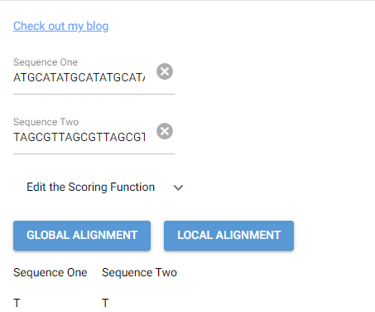
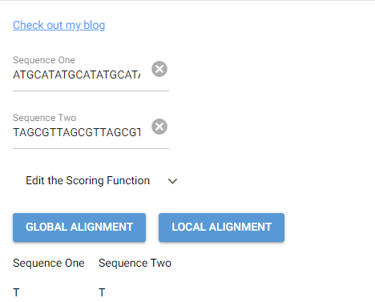
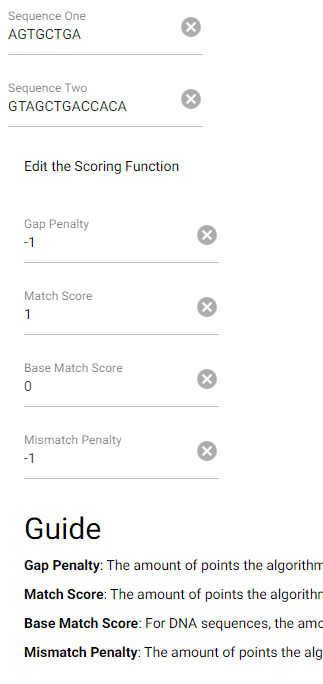
P. S. I broke up the UI into images