

Global and Local Alignment Explained
Global and local alignment are essential for comparing DNA, RNA, or protein sequences. Furthermore, these two techniques are important to understanding more advanced methods in genomics, such as the BLAST database search and multiple sequence alignment.
GENOMICSEXPLAINERBEGINNER FRIENDLY
Sohum Bhardwaj
8/7/20246 min read
Even though Sequence Alignment is a relatively simple technique, it is extremely useful to scientists that work with sequences across many fields
For example, evolutionary biologists want to figure out the relationship between different species and seek to figure out whether they share a common ancestor or not. A common ancestor is an organism that both species share as an ancestor . If scientists can identify that two species do have a common ancestor, they can get insights into how the two species diverged and specialized for their niches (role in the environment) or learn more about the common ancestor of the species. Scientists use alignment techniques to find similarities in biological sequences like DNA which can provide evidence of a common ancestor. Giant genomics databases exist that scientists use to lookup sequences that are similar to the one that they are testing in order to find these relationships.
Another example of the usefulness of alignment is within functional genomics. Sequences that are similar likely have a similar function. Functional genomics scientists can take newly sequenced genes and find similarities to already sequenced ones through sequence alignment. This can help them infer the purpose of these genes. Of course the same can be done for amino acid sequences to infer the purpose of the protein. This helps functional genomics scientists understand the purpose of the gene and provide context to how it interacts with regulatory systems.
Premise of Sequence Alignment
Sequence Alignment is a technique that biologists use to compare sequences. Consider the following "sequences":
BIOLOGYISPRETTYCOOL
PRETTYCOOLRIGHT
If you were asked to find the similarity between these two sequences of characters, you may highlight that these sequences both contain the phrase "PRETTYCOOL":
BIOLOGYISPRETTYCOOL
PRETTYCOOLRIGHT
This is essentially what these algorithms are doing except with one more twist. We can add gaps or spaces to find even better alignments
THISISAREALLYLONGSEQUENCE
THISISALONGSEQUENCE
With gaps we might align this to:
THISISAREALLYLONGSEQUENCE
THISISA - - - - - - LONGSEQUENCE
Notice how the gaps are denoted by dashes. In terms of an actual biological sequence gaps can represent point mutations like insertions or deletions which is when an additional base is inserted into a DNA sequence by an error of DNA polymerase or when one is removed.
This is the process of global alignment, or aligning a sequence from end to end. Another tool that biologists use is local alignment where similar parts of a sequence are aligned. This is depicted below. The similar part of sequence S and T is matched but the rest of either sequence is not used
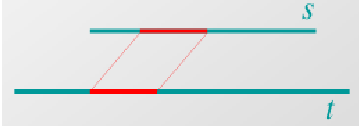
Devising an Algorithm for Sequence Alignment
Sequence alignment looks like a task that an algorithm can do but first we should define the problem in terms of something an algorithm can fulfill:
Sequence Alignment Problem:
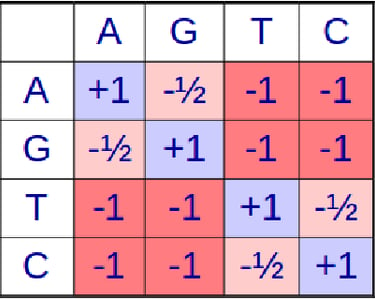
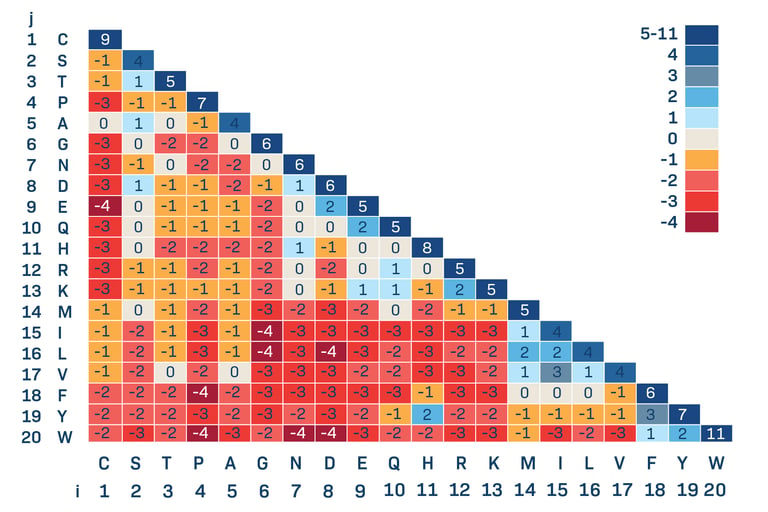
Given sequence S and sequence T find the best alignment (highest score) of the two sequences according to a scoring matrix (see sample scoring matrices on the right) and gap penalty G
Understanding the Scoring Matrix
The intersection of a column and a row in the scoring matrix shows the score for the alignment of those two bases or amino acids. A negative score indicates that the alignment does not signify a relationship between the two sequences and positive score means an alignment does provide evidence for a relationship of two sequences.
Gap Penalty
A gap penalty is the amount of points deducted for each gap in the sequence. Usually more than the mismatch penalty which is the points deducted for an incorrect match.
Needleman-Wunsch Algorithm
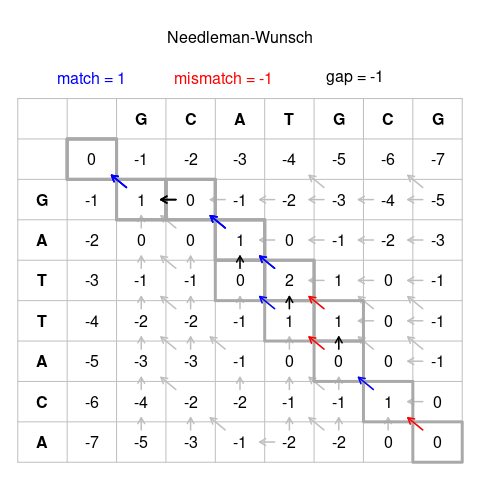
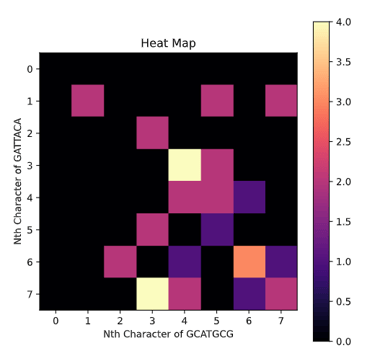
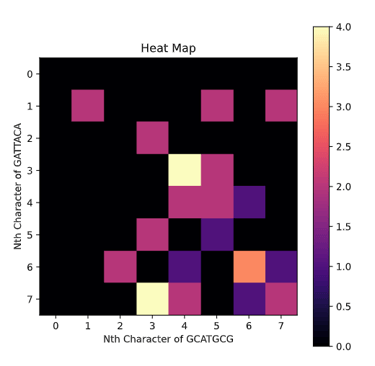
The Needleman-Wunsch algorithm is an important algorithm for Global alignment and it works by using a programming technique called dynamic programming. Both sequence S and T used to construct a matrix with one being the y-axis and the other being the x-axis. Lets say the S is GCATGCG and T is GATTACA.
Now each box of the matrix represents the pairing of the ith character of sequence S and the ith character of sequence T. First we will fill in all of these boxes.
The top row and leftmost column are what happens when we pair the ith character of a sequence to a gap so box (0, 0) would be filled as 0 and box (0, 1) would be filled out as the previous box minus the gap penalty and so on. For this case the gap penalty is -1.
Now we reach box (1, 1) where we have around 3 options.
1. We pair S(1) with T(1), or G with G by extending the box(0,0)
2. We pair a Gap with T(1), or - with G by extending box (0, 1)
3. We pair a Gap with S(1), or G with - by extending box (1, 0)
There is a clear winner here: pairing S(1) with T(1). We add the score for a direct match, +1 with the score of box(1, 1) with is 0 to get the score of box(1, 1) as 1. Notice how we extend from previous boxes instead of calculating the best options all over again. Each box represents the score of the best possible alignment that ends with S(i) paired with T(j) for box (i, j). With box (2, 1) it is
1. We pair S(2) with T(1), or C with G by extending the box(1,0)
2. We pair a Gap with S(1), or C with - by extending box (1,1)
3. We pair a Gap with T(1), or - with G by extending box (2, 0)
The best option here is option 2. In Option 1, box(1, 0) holds the score of -1 and matching C with G is a mismatch which carries an additional score of -1 bringing the total score to -2. Option 3 is worse as it results in a score of -3. Option 2 however results in a score of 0 as box (1, 1) has a score of 1 which is subtracted by the gap penalty of 1 to get 0.
These options can be generalized however to these three:
1. We pair S(i) with T(j), lookup and add the score from the score of S(i) and T(j) to the sequence
2. We pair a Gap with T(i), pay the gap penalty
3. We pair a Gap with S(j), pay the gap penalty
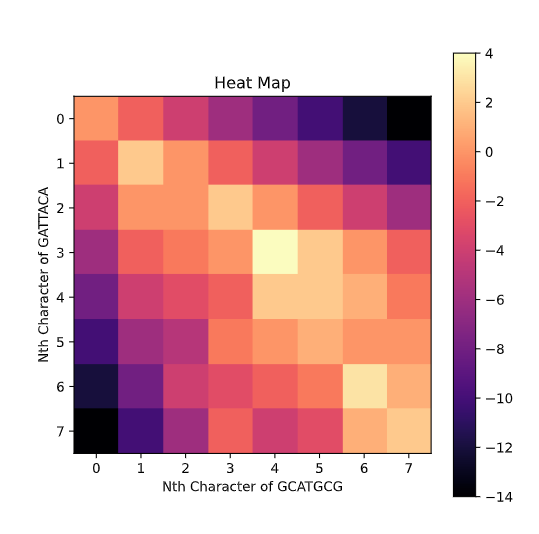
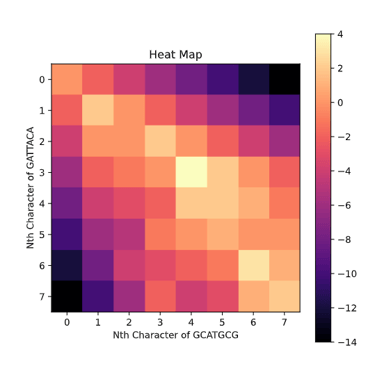
While we fill out the matrix, it is important to keep track of which box was extended. This is because once the matrix is filled out we can trace back from the bottom right box which represents the score of the best end-to-end alignment. The arrows represent this traceback.
Smith-Waterman Algorithm
This algorithm is very similar to the Needleman-Wunsch algorithm except it is for Local Alignment instead of Global alignment. There are a few modifications that we need to make to NWA to make it suitable for local alignments. Firstly, for a local Alignment we want to match the most similar parts of each sequence no matter where they are. Therefore if a box would be negative want to set it to 0 so that a sequence can potential be extended from it without suffering any penalties. We do this by adding an option 4:
1. We pair S(i) with T(j), lookup and add the score from the score matrix of S(i) and T(j) to the sequence
2. We pair a Gap with T(i), pay the gap penalty
3. We pair a Gap with S(j), pay the gap penalty
4. 0
Yes, the option is literally just to take a score of 0 and set the box to equal 0. This means if option 1-3 are all negative we will have the box set to 0. This will cause a lot of mini-alignments to spring up from like portions of the sequence and then fizzle out into 0's. In order to pick the best one of these local alignments we find the highest score in any box and trace it back until we reach a 0 at which we terminate and return the highest scoring alignment. If we run this algorithm on S and T we get
GCAT
G--AT
Notice how in the Heat Map representation most of the area is black indicating 0's
Why Sequence Alignment matters
Sources
Code Used Here
I implemented both of these algorithms and created a UI that you can use to enter sequences, configure the gap penalties and score matrix, and view the alignment as well as a heat map showing how the algorithm found the alignment. Check it out on my github page.